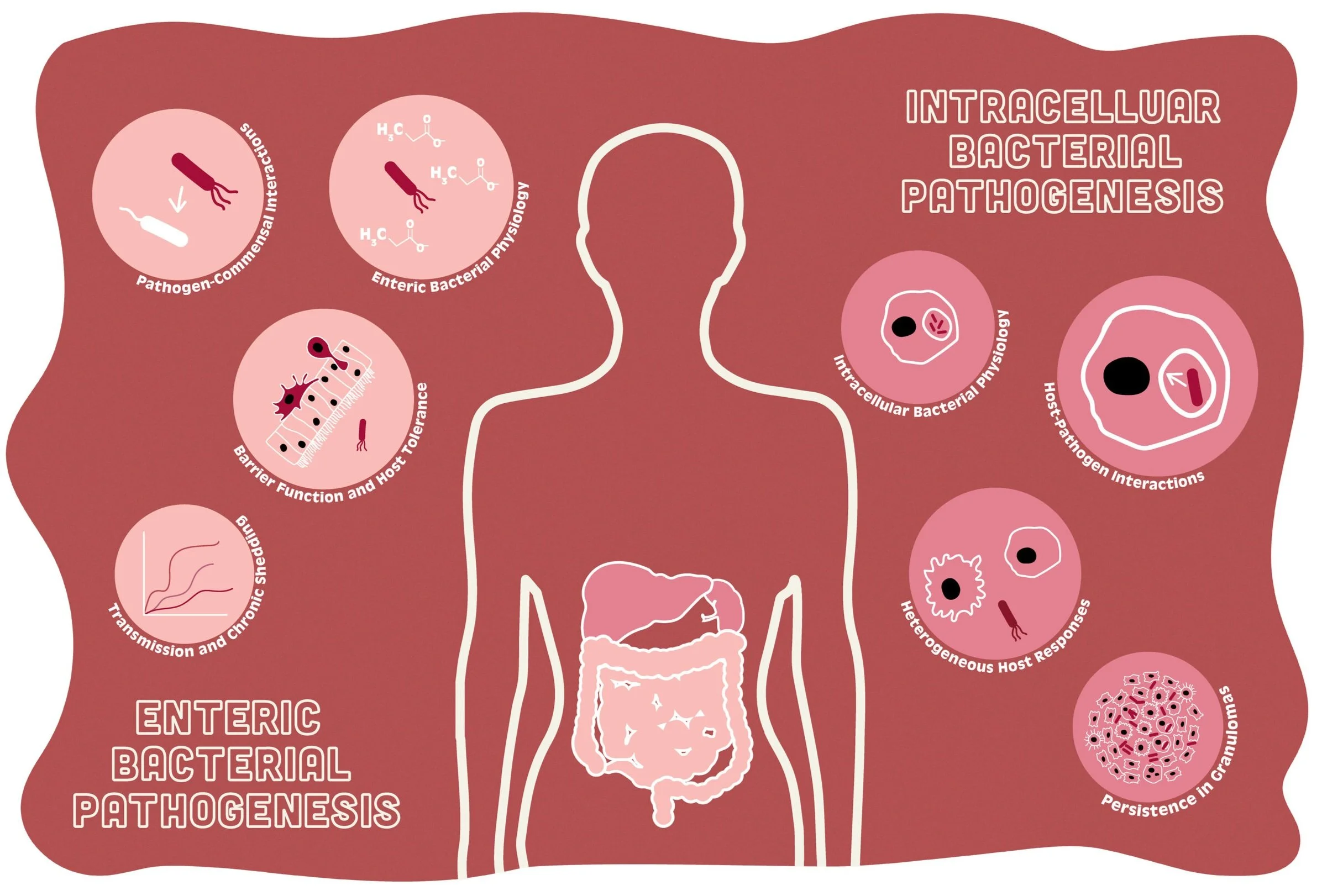
Jacobson et. al., Cell Host & Microbe 2018
Lam and Monack, PLoS Pathog 2014
Sana et. al., PNAS 2016
Lawley et. al., Infect Immun 2008
Co et. al. Cell Reports 2019
Gopinath et. al., PLoS Pathog 2013
Gopinath et. al., PNAS 2014
Eisele, Ruby et. al. Cell Host & Microbe 2013
Pham et. al., Cell Host & Microbe 2020
Carden et. al., Cell Host & Microbe 2017
Broz et. al., Nature 2012
McLaughlin et. al., PLoS Pathog 2009
Lawley et. al., PLoS Pathog 2006
Napier et. al., PNAS 2019
Lane et. al., Cell Systems 2019
Napier et. al., J Exp Med 2016